One of the most popular RNA-seq mappers is TopHat which aligns reads in two steps. Classroom Guide for Students.
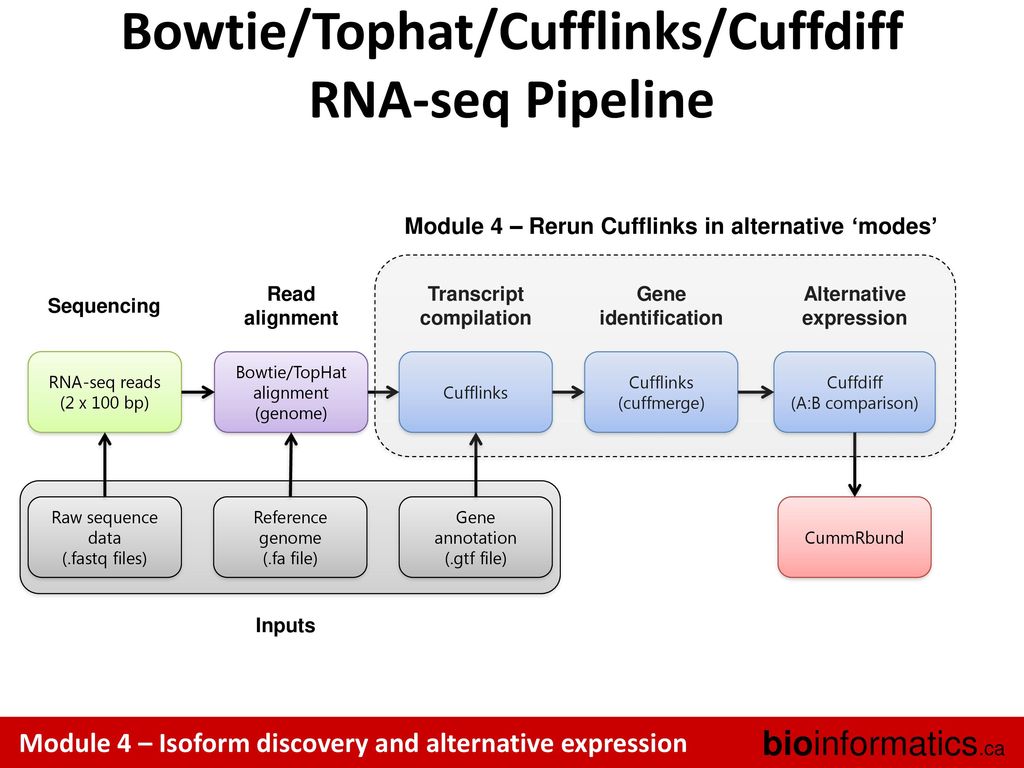
Canadian Bioinformatics Workshops Ppt Download
Principles of transcriptome analysis and gene expression quantification.

. 10182012 Release of MATS 304beta Fixed a bug related to gene expression level calculation. RNA Analysis Tophat tool to map RNA-seq reads to the hg19 build. 10122012 Release of MATS 303beta Fixed a bug related to gene expression level calculation.
BAM files from TopHat 2xx or Bowtie 2xx now work properly. Look at requested time accuracy using XDMoD. There are many bioinformatics tools available to perform the alignment of short reads.
Data created by different labs can have features that make it difficult to have an single analysis technique. Unspliced reads are mapped to locate exons with Bowtie. Featured RNA-Seq Job Staff Scientist Single Cell RNA-Seq and Bioinformatics 12 days ago Leave a comment 396 Views The Biocenter of the University of Würzburg is an interdisciplinary center for basic research with currently 14 departments and chairs.
Dimensionality reduction and clustering Loupe Cell Browser 10X Genomics. Thus a single short read might align to two locations that are separated by 10000 bp. Use the NGS.
Classroom Project Resource Guide Toggle submenu visibility. 比如2019年06月发表在SR的文章Systematic identification and characterization of Aedes aegypti long noncoding RNAs lncRNAs 就是一个比较新而且比较容易学习的鉴定. Annovar bioconductor bowtie bwt CHIP-seq ENSEMBL GEO GSEA limma linux miRNA-seq mutation mysql ncbi Peak perl R RNA-seq samtools shell snp SRA TCGA UCSC vcf 包 变异 可视化 基因组 差异分析 差异基因 数据库 文献 服务器 模块 比对 注释 癌症 直播 突变 脚本 芯片 转录组 转.
While our RNA-seq simulations were useful for defining an unambiguous truth set and evaluating the impact of fusion expression levels and read length some characteristics of real RNA-seq data are not presently modeled such as reverse transcription artifacts and off-target transcription eg non-spliced introns and intergenic transcription. Instead it is better to use a mapper such as Tophat that is designed to map RNA-seq reads. We also have provided an example below showing how to build and install your own Python packages and make them available inside of Python.
Using nbgrader for Classroom. RNA-seq mappers need to solve an additional problem not encountered in DNA-only alignment. Add Python packages using the conda.
You can change this number by clicking on System category. Many RNA-seq reads will span introns. This is the average distance in basepairs between reads not the total insertfragment size.
Because the reads are paired youll need to set mean inner distance between pairs. In this case I change the number of CPUs to 4 since 4 is the largest value shown on the green bar in my case. Since Tophat program can take an advantage of multiple processorsthreads it is a good idea to specify a large number of processors in virtual machine default value is 1.
火山3D volcano3D软件包可用于探索三组之间差异表达的探针 其主要目的是在三维火山图中可视化差异表达的基因 这些图可以使用图转换为交互式可视化 该插图探讨. If it is a commonly used package or one that is particularly difficult to compile you can contact OSC Help for assistance. Unmapped reads are then split and aligned independently to identify exon junctions The RNA-seq read alignment program.
For reads with multiple alignments to store the alignment score for the next-best. Using Rstudio for classroom. XS for example is used by TopHat an RNA-seq analysis tool we will discuss later to encode the strand information eg XSA while Bowtie2 and BWA use XSi.
Raw read alignment QC and matrix generation for scRNA-seq and ATAC-seq. HOWTO Toggle submenu visibility. 8302012 Release of MATS 302beta Fixed a bug related to default insert sizes and standard distribution.
Traditional RNA-Seq tools mainly focus on mRNA which has different features than GRO-Seq and are generally not useful for identifying GRO-Seq transcripts. Just as with ChIP-Seq not all GRO-Seq data was created equally. While we provide a number of Python packages you may need a package we do not provide.
The RNA sequencing processing captures and sequences mature messenger RNA molecules from which introns have been removed by splicing. Now you can click OK button to continue. Using Jupyter for Classroom.
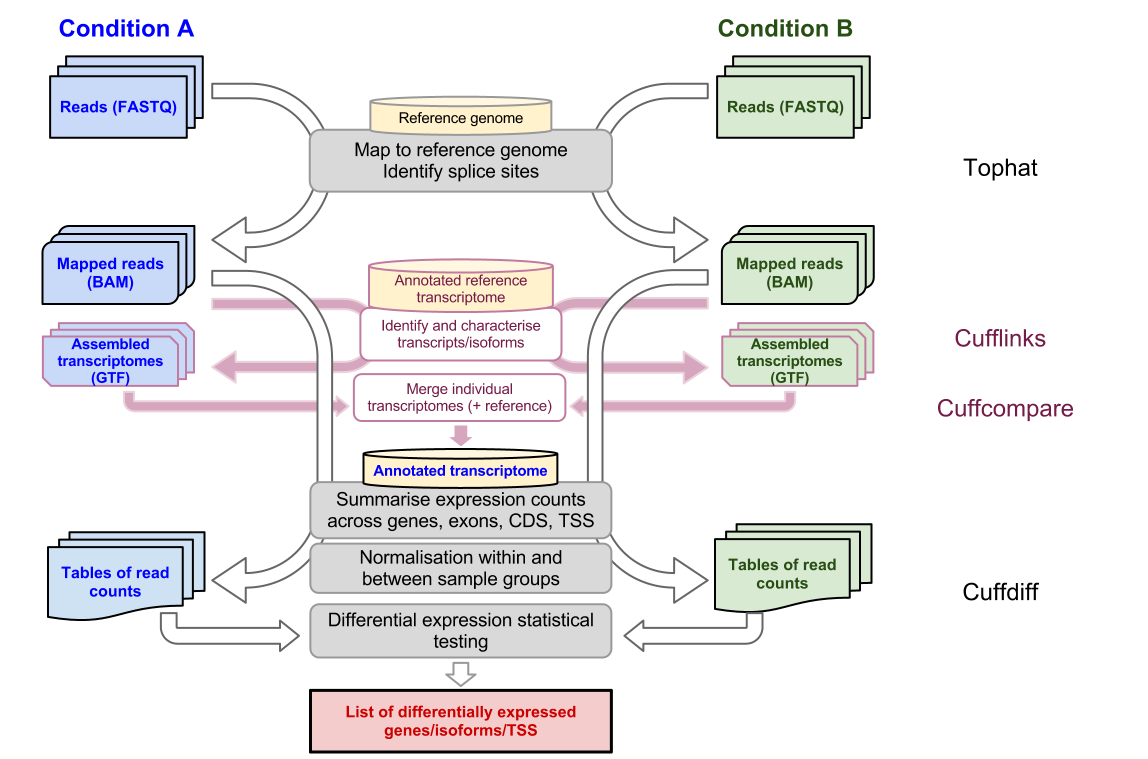
Introduction To Bulk Rnaseq Analysis Bioinformatics Documentation
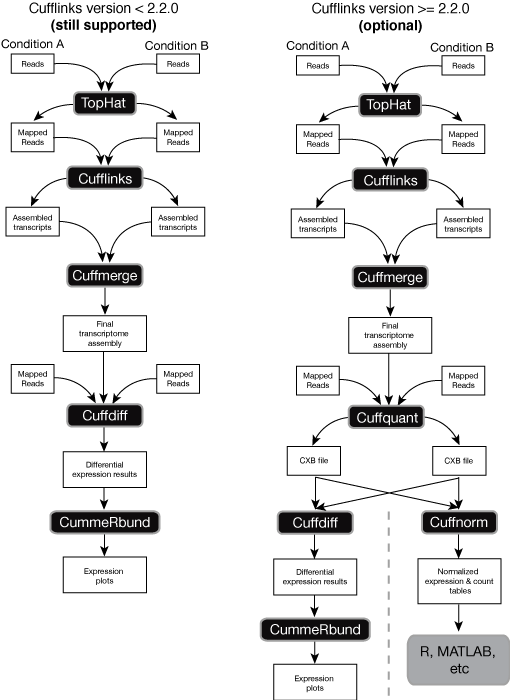
The Cufflinks Rna Seq Workflow

Tophat Cufflinks Command Pipeline

Reference Based Rnaseq Data Analysis Long
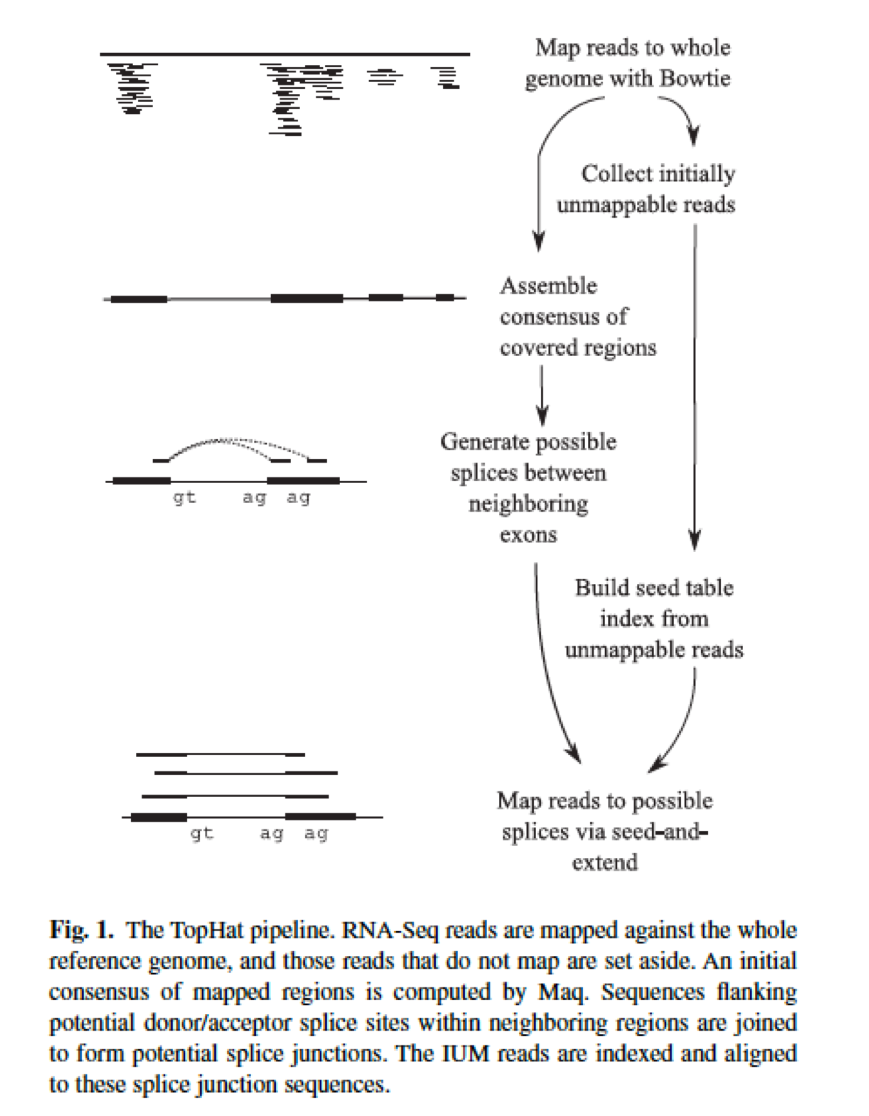
Aligning Rna Seq Data Ngs Analysis
Incorporating Rnaseq Tophat To Augustus Computational Biology

Basic Analyses With Tophat Cufflinks Rnaseq Tutorial 1 Documentation

Rna Seq Alignment And Visualization Youtube
0 comments
Post a Comment